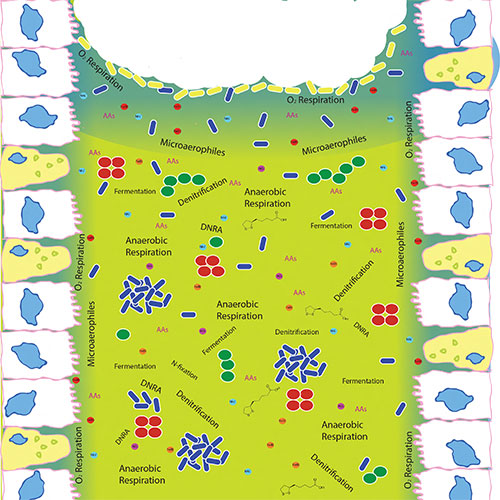
A mucus filled bronchiole lined with epithelial cells has an inherent oxygen gradient
(blue), but is mostly anaerobic (green). This oxygen gradient stratifies and drives much
of the microbial physiology. The image shows microbial metabolisms and various nutrients
as determined through analysis of metagenomic sequence data and metabolomics.
We are dissecting how microbial and viral communities persist in the lungs of people with
cystic fibrosis (CF) in order to design new treatment strategies. We use a metagenomic
approach to characterize the communities, and we collaborate with a range of clinicians,
bioinformaticians, chemists, engineers and mathematicians to generate samples, data and
ideas. In collaboration with Dr. Douglas Conrad who runs the adult CF clinic at UCSD, we
collect sputum samples from patients over time.
The lives of people with cystic fibrosis are punctuated by flare ups of the chronic
infections and inflammation in their lungs, which are normally treated with antibiotics.
We take sputum samples before, during and after these flare ups and examine the
microbial and viral communities through metagenomic sequencing of their DNA and RNA.
Once or twice a year we also obtain an explant lung when a patient undergoes a lung
transplant.
Knowing both the genetic potential and real-time expression of genes responsible for
different types of metabolism throughout the treatment cycle is helping us understand
the lifestyle and survival strategies of the organisms in these communities. This may
enable development of microbial biomarkers of disease state that predict an oncoming
flare up before the inflammation and damage become full blown.
A surprisingly large number of patients in this San Diego population are colonized by Rothia
mucilaginosa, which is not typically included in clinical laboratory characterizations
of CF lung infections. One consistent theme that is apparent in all of our results is that each
CF patient harbors a unique microbial community, comprised of different proportions of bacterial
taxa with specific sub-species and strains that have assembled and evolved under the conditions
of a particular lung. The lack of clear signatures of disease state across patients is an
important reminder that one way to improve medical control of CF lung infections is to design
individual treatments that interfere with a particular mode of survival.
In collaboration with Sara Zarei and Peter Salamon in the SDSU
biomath group, we are comparing MRI data from CF patients with models of the demise of lung
function as increasing numbers of lung tube branches are plugged and irreversibly damaged. We
are also analyzing breath gas samples from CF patients with the Rowland-Blake group at UCI to
characterize the volatile molecules present in their breath, and comparing them with healthy
patients and background room samples. Early results include molecules that indicate synergisms
between Streptococcus spp. and phenazine producing microbes such
asPseudomonas, which provide alternative electron acceptors and enable anaerobic
respiration. To test the synergisms we identify through metagenome and metabolome analysis, we
are establishing systems for culturing CF lung-derived organisms in conditions that mimic the CF
lung. Establishing model systems for CF-associated microbial communities will also allow us to
test how novel treatments such as hyperbaric oxygen affect microbial community structure and
function.
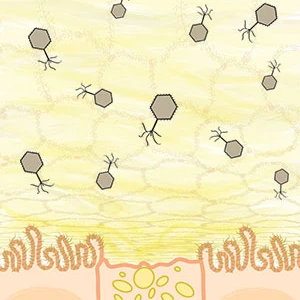
The protective layer of mucus on the body's surface serves both as an
entry point for pathogens and a home for large populations of beneficial microbes. This mucus
layer harbors a large diversity of both bacteria and phage. We show elevated concentrations of
phage on all mucosal surfaces sampled, ranging from cnidarians to humans, compared to the
surrounding environment.
Using bacteriophage T4 and various in vitro tissue culture cells as a model system, we
demonstrate that this increase in phage abundance is mucus-dependent. This phage-mucus
association reduces bacterial attachment and colonization of the mucus, which subsequently
protects the underlying epithelium from bacterial infection. Enrichment of phage in mucus occurs
via binding interactions between variable glycan residues displayed in mucus and
immunoglobulin-like protein domains exposed on phage capsids.
Based on these observations we propose the Bacteriophage Adherence to Mucus (BAM) model that
provides a ubiquitous, but non-host-derived, immunity applicable to mucosal surfaces. This
benefits the metazoan host by limiting mucosal bacteria, and benefits the phage through more
frequent interactions with bacterial hosts. BAM suggests the first demonstration of a symbiotic
interaction between phage and metazoan hosts that provides a previously unrecognized immunity
that actively protects mucosal surfaces.
Viruses, and bacteriophage in particular, are the most diverse and numerous biological entities
on the planet. Phage interactions with bacterial hosts, which can be observed through population
size modulation by outright killing or possessing ecologically relevant genes conferring host
growth advantage, have a profound influence on environmental nutrient cycles. Unfortunately, how
phage influence community dynamics is poorly understood due to the fact that only 10 to 30% of
viral genes found in the environment have sequence similarity to any other identified protein,
let alone any protein of known function. The goal of this project is to characterize these phage
proteins of unknown function using a combination of high throughput computational and
physiological methods, in concert with protein crystallography, to gain insight into phage
biodiversity, along with the environmental forces driving phage evolution, and develop a better
understanding of host-phage interactions.
Collaborators: Dr. Anca Segall, Dr. Rob Edwards

Cnidarians diverged from bilateria 550 million years ago and are considered to be the basal phyla
to all metazoan life. Recent work has suggested that despite their morphological simplicity, the
cnidarian immune repertoire is highly complex with many unexpected similarities to the human
immune system (see references). To investigate the cnidarian immune response we utilize
molecular tools developed for the human system and apply them to reef building corals as well
freshwater Hydra species. In addition to functionally characterizing the cnidarian immune system
we are investigating how the resident viral population interacts with host immune components to
create a predicated map of the coral immune response. Taken together this project provides novel
insight into the general evolution of immunity with potential application to human biology.
- Putnam et al., 2007
- Shinzato et al., 2011